Besides its catalytic function in the TCA cycle, the Aconitase enzyme has an
important regulatory function in both Eukaryotes and Prokaryotes. When
active as a regulator, Aconitase binds specific structural motifs in mRNA,
so-called Iron-Response Elements (IREs). In Eukaryotes, it is highly
sensitive to iron deprivation and oxidative stress. In bacteria, the
role of Aconitase seems to be similar, though the recognition sequence
is less preserved. Aconitase recognises the RNA stem loop structure of
the IRE consisting of a C bulge, a stem of 6 amino acids, and a loop of 5 or
6 nucleotides. In Eukaryotes, the loop sequence always is CAGUG, in
Bacteria, more variations are possible.
In this example, we will build a set of patterns to identify putative IREs
in the 5’ UTR sequences of the bacterium Streptomyces coelicolor. First
starting with a relatively basic example, we will slowly extend it to cover
more advanced patterns and use cases.
The basic IRE pattern is
CN1N2N3N4N5N6CAGUGN'6N'5N'4N'3N'2N'1,
where Nx is any nucleotide, and N'x is its complement.
An initial attempt at finding such a pattern using PatScan would use a number of string patterns and a
complement pattern. Just using a string pattern with CNNNNNNCAGUGNNNNNN would fail to ensure the second
set of Ns would be the reverse complement of the first, so instead we need to break the pattern up a bit.
example1_input.fa file.
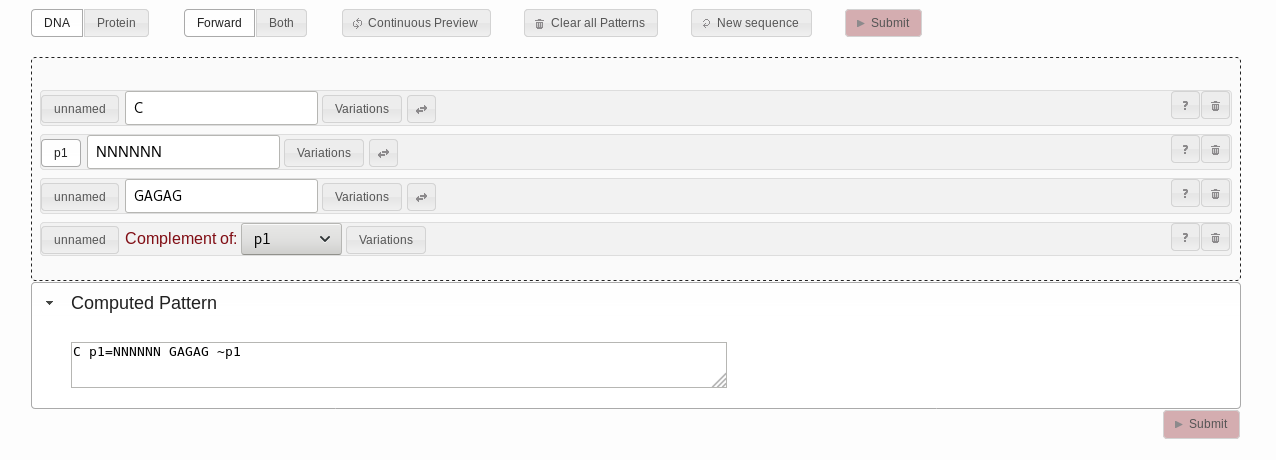
C. For the second string pattern,
select NNNNNN. For the third string pattern, select
CAGUG. Click the "unnamed" button next to the second string
pattern. The button text will change to "p1", the identifier of this
pattern. In the complement pattern, you can now select the “p1”
identifier for the “complement of” dropdown. Once you are done, your
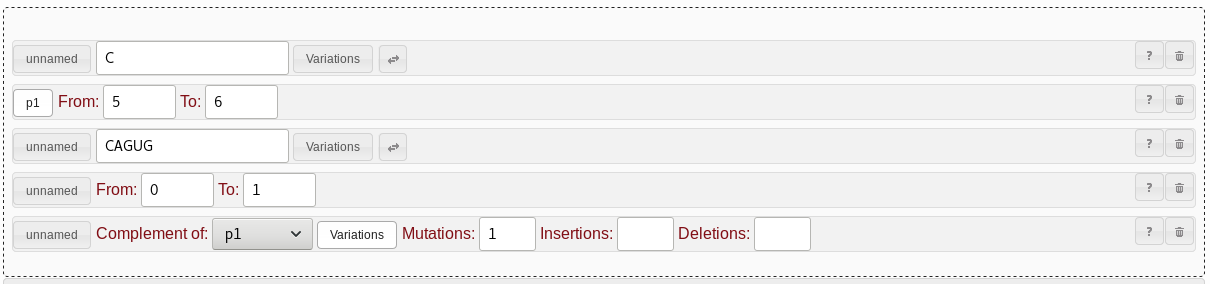
work area should look like in Fig. 4.
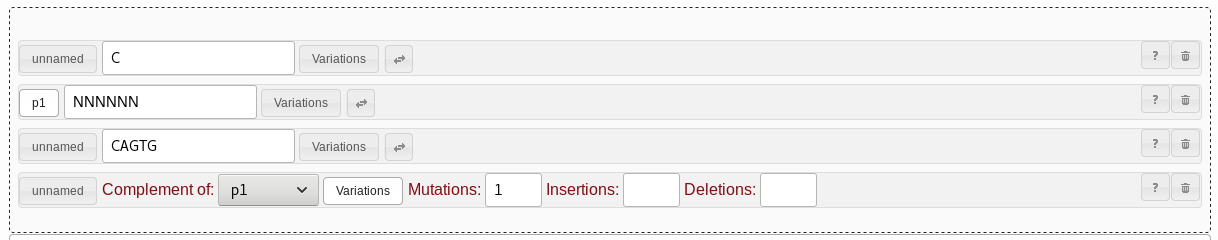
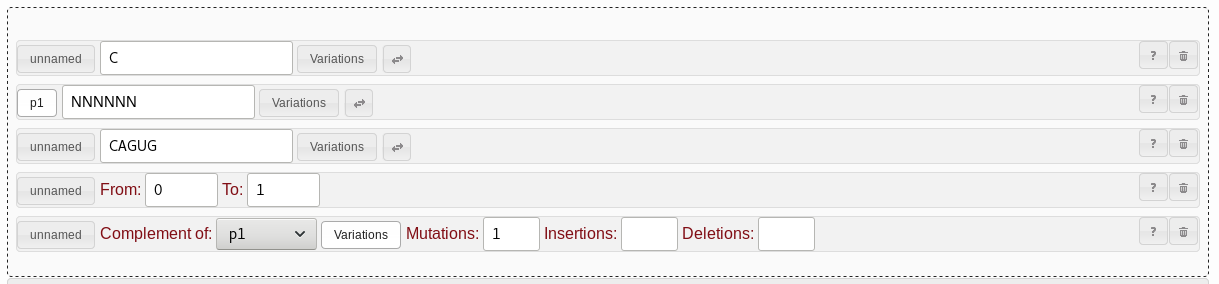
CAGUG pattern. Let's add support for this by adding a range pattern. Drag and drop
the range pattern field to sit between the string pattern for the loop
region and the repeat pattern for the reverse part of the stem loop.
Set the range pattern to cover from "0" to "1" bases.
NNNNNN string pattern, and drag another range pattern into
the same location instead. Remember to hit the "unnamed" button again so
it says "p1" as before. You also need to select that from the
"Complement of" dropdown of the complement pattern again.
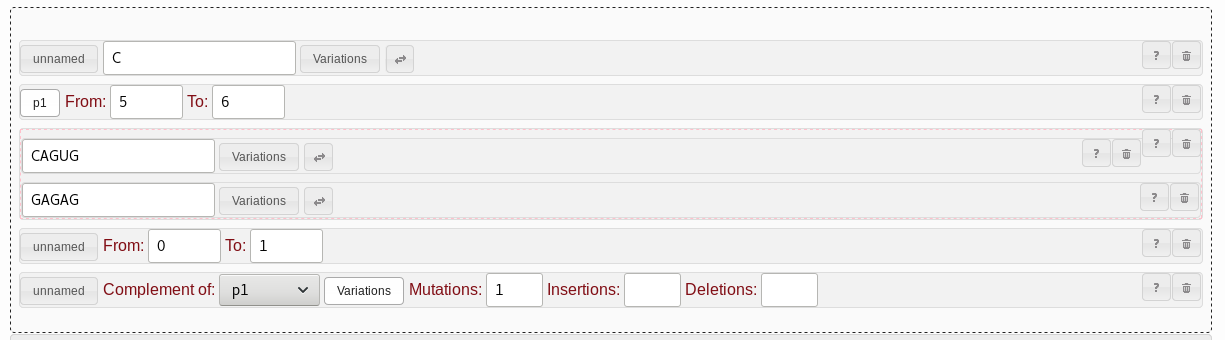
GAGAG has also been
reported to be functional. To support searching for CAGUG
or GAGAG at the same time, we can use the
alternative pattern.
It is basically a container that can hold any two other patterns, one of
which has to match. To add it, drag it into position next to the
CAGUG string pattern, and then drag the CAGUG
pattern into the alternative pattern. Drag a new string pattern into the
remaining free slot of the alternative pattern, and set that sequence to
GAGAG. It does not matter what order the two string patterns
have inside the alternative pattern.
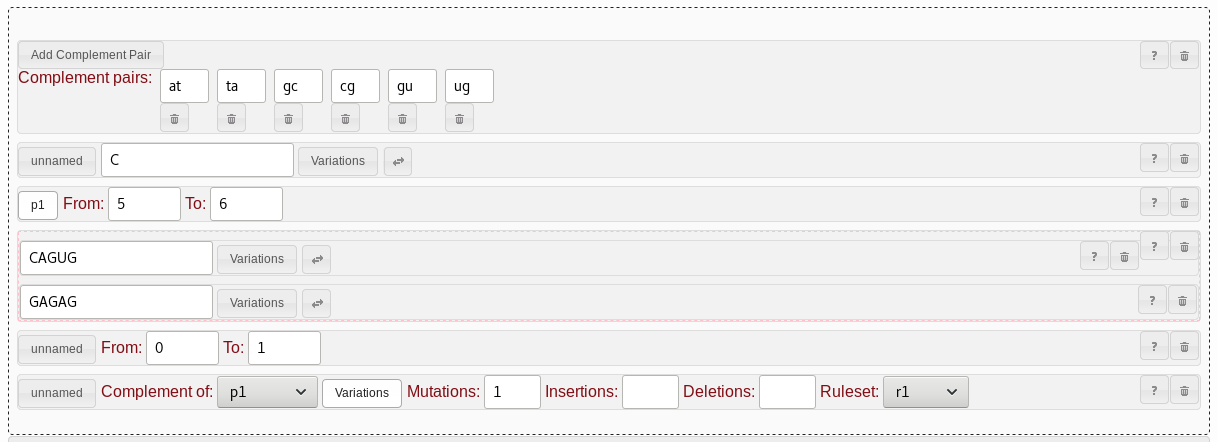
GU/UG pairs are possible.
To support this, you can create custom
alternative
complementation rules. Note that due to constraints in PatScan,
the alternative complementation rules need to be specified before being
usable in a complement
rule. Personally, I prefer having all alternative complementation
rules at the start of my pattern set.GU pair does not set up pairing
rules for UG.
CAGUG and GAGAG but also want to allow for a
G base in the fourth position, we would use the following
approach:C or G, so in the first column, we fill in
"50" in the second and third rows. Zeroes can be left out, so no need to
fill out the other rows. The second base should always be an
A, so we put "100" into the first row of the second column
and leave the rest blank. The third base should always be a
G, so "100" goes into the third row of the third column.
The fourth base could be A, G, or
T (actually U, but for weight matrices those
bases are equivalent). The weights are a bit arbitrary for this one, as
long as C gets a zero. As there is no published evidence
for G actualy being valid, I personally like to score this
a bit less than the experimentally validated options. So, for column
four, let's put "40" into the first and last rows, and "20" into the
third row. For the last base, we again always want a G, so
the fifth column gets a "100" in the third row.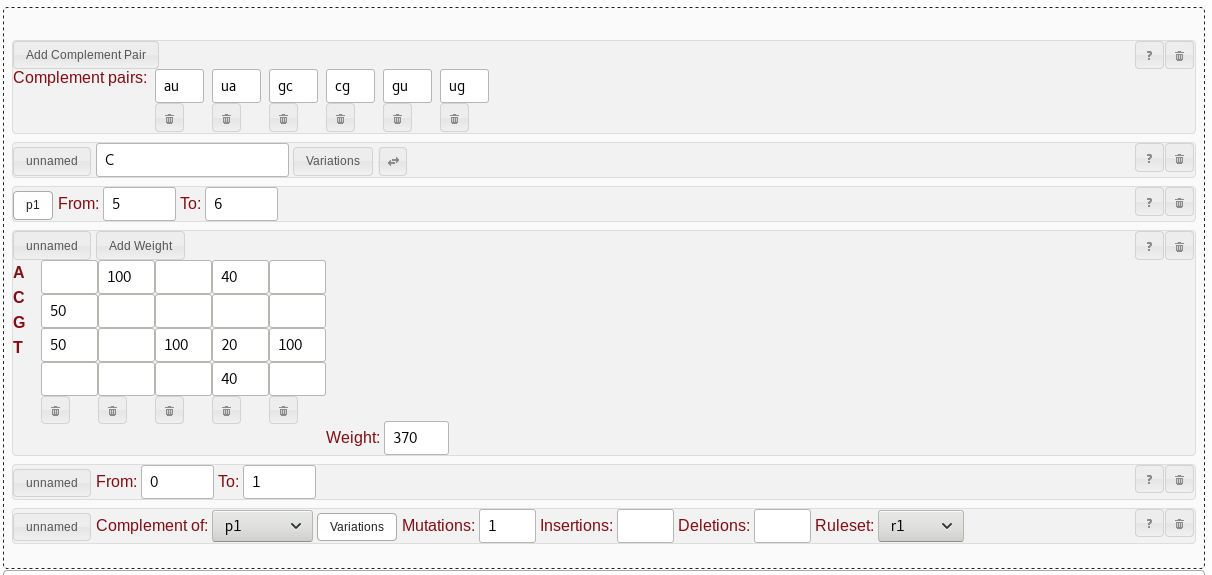
Cytochrome P450 proteins utilise heme cofactors to perform oxidation reactions and are found all over the tree of life. The heme cofactor binds to a cysteine in the active site of the enzyme. In this example, we will build a set of patterns to identify the active site cysteine of cytochrome P450 enzymes found in the proteome of the bacterium Streptomyces coelicolor. We assume that you have already gone over example 1 and are familiar with the basic pattern creation.
According to the PROSITE database entry PS00086,
the pattern for identifying the active site cysteine is
[FW]-[SGNH]-x-[GD]-{F}-[RKHPT]-{P}-C-[LIVMFAP]-[GAD]. If you
are not familiar with the PROSITE pattern notation, amino acid letters
wrapped in square brackets ([ ]) are alternatives, amino
acid letters wrapped by curly braces ({ }) should not be present
at a given position, and x means any arbirary amino acid.
[ ]) in the PROSITE format. The
not-any-of pattern allows to
give a list of amino acids that should not be present, matching the curly braces ({ })
in the PROSITE format. Note that both the any-of and the not-any-of pattern
don't take a location but instead need to be inserted into your overall pattern
at the location where you want to allow/disallow multiple amino acids.
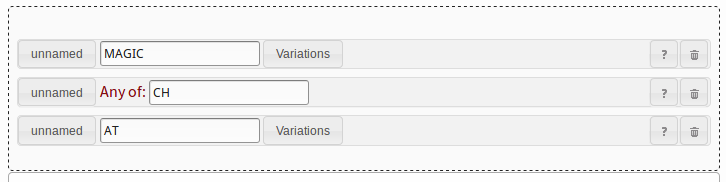
So if you wanted to match the sequences MAGICHAT and MAGICCAT,
you want to allow for both H and C at position 6 in the sequence.
In order to build this pattern in PatScan, you need to break it up into three parts.
You need a string pattern for MAGIC, an any-of pattern for CH
and another string pattern for AT, as shown in figure 11.
The PROSITE pattern for this would be MAGIC-[CH]-AT, so you can usually
use the dashes (-) in the PROSITE pattern to figure out where
you need differnt pattern elements.
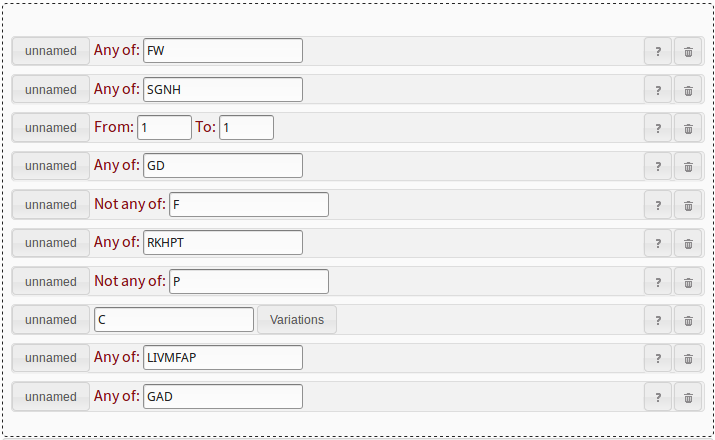
[FW]-[SGNH]-x-[GD]-{F}-[RKHPT]-{P}-C-[LIVMFAP]-[GAD]. We will
need ten pattern elements. The fastest way to add them is to click on
the respective elements in the selector. You will need two any-of patterns,
a range pattern, an any-of pattern, a not-any-of pattern, an any-of pattern,
a not-any-of pattern, a string pattern, and two any-of patterns. The first
two any-of patterns take FW and SGNH, respectively.
There is a single x in the PROSITE pattern, which translates to
a range pattern from 1 to 1. The next any-of pattern
takes the GD. The not-any-of pattern takes an F.
The next any-of pattern takes RKHPT, the not-any-of pattern takes
a P. The single C goes into the string pattern.
The last two any-of patterns take LIVMFAP and GAD,
respectively. See figure 12 for the example.
Clicking "Submit" sends off the request, yielding eleven hits
on the Streptomyces coelicolor proteome.